Annotating human BMMCs#
Annotate a dataset of human bonemarrow mononuclear cells (BMMCs) [LBC+21]. This data corresponds to the gene expression portion of the NeurIPS 2021 challenge 10x multiome data.
The original raw version of this dataset can be obtained from GEO. Running this notebook will automatically download the data from figshare.
Preliminaries#
Import packages & data#
import scanpy as sc
import matplotlib.pyplot as plt
import seaborn as sns
from cell_annotator import CellAnnotator, ObsBeautifier
from cell_annotator.utils import _shuffle_cluster_key_categories_within_sample
Load the gene expression portion of the NeurIPS 2021 challenge 10x multiome data. This dataset has been obtained from GEO. We computed an embedding using scANVI to visualize the data [XLM+21].
adata = sc.read("data/human_bmmcs.h5ad", backup_url="https://figshare.com/ndownloader/files/51994907")
adata
AnnData object with n_obs × n_vars = 69249 × 13431
obs: 'pct_counts_mt', 'n_counts', 'n_genes', 'size_factors', 'phase', 'cell_type', 'batch', 'pseudotime_order', 'Samplename', 'Site', 'DonorNumber', 'Modality', 'VendorLot', 'DonorID', 'DonorAge', 'DonorBMI', 'DonorBloodType', 'DonorRace', 'Ethnicity', 'DonorGender', 'QCMeds', 'DonorSmoker', '_scvi_batch', '_scvi_labels'
var: 'feature_types', 'gene_id'
uns: 'DonorNumber_colors', 'Samplename_colors', 'Site_colors', '_scvi_manager_uuid', '_scvi_uuid', 'batch_colors', 'cell_type_colors', 'dataset_id', 'genome', 'neighbors', 'organism', 'phase_colors', 'umap'
obsm: 'X_pca', 'X_scANVI', 'X_umap', '_scvi_extra_categorical_covs'
layers: 'counts'
obsp: 'connectivities', 'distances'
Preprocess the data#
Create numerical cell type labels to be used for annotation and a shuffled version thereof.
adata.obs["leiden"] = adata.obs["cell_type"].copy()
adata.obs["leiden"] = adata.obs["leiden"].cat.rename_categories(
{cat: str(i) for i, cat in enumerate(adata.obs["cell_type"].cat.categories)}
)
adata = _shuffle_cluster_key_categories_within_sample(adata, sample_key="Site", key_added="leiden_shuffled")
Visualize
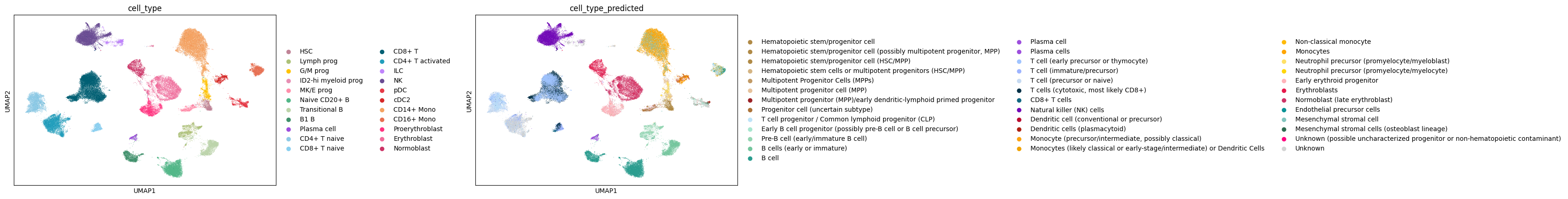
sc.pl.embedding(adata, basis="umap", color=["Site", "DonorNumber", "cell_type", "leiden_shuffled"], wspace=0.7, ncols=5)
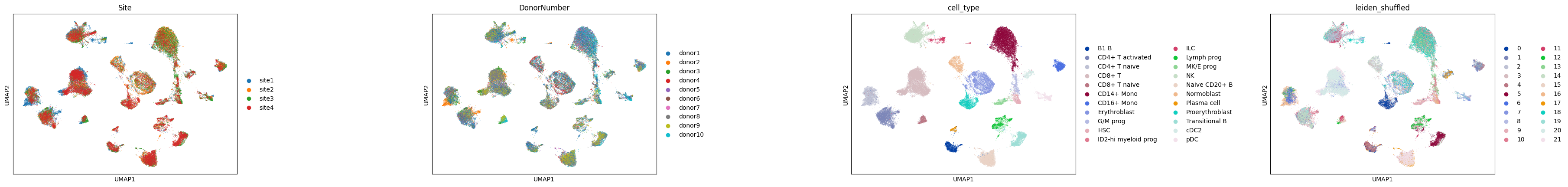
We’re using the Site annotation to denote different samples, and we randomly shuffled the order of cluster labels within each sample to resemble a realistic scenario, in which we’re given a dataset with several samples, with independent clustering per sample (i.e., pre-integration).
Query cell type labels per sample#
In this notebook, we will demonstrate the slightly more advanced usage of this package, which involves a few method calls, but gives you more control and insights.
If you’re new and looking for the simplest way to get started, head over to our basic tutorial: Annotating the heart atlas.
cell_ann = CellAnnotator(
adata,
species="human",
tissue="bone marrow mononuclear cells",
cluster_key="leiden_shuffled",
sample_key="Site",
model="gpt-4.1",
)
cell_ann
INFO ✅ OPENAI API key is available
INFO Initializing `4` SampleAnnotator objects(s).
🧬 CellAnnotator
================
📋 Species: human
🔬 Tissue: bone marrow mononuclear cells
⏳ Stage: adult
🔗 Cluster key: leiden_shuffled
🔬 Sample key: Site
🤖 Provider: openai
🧠 Model: gpt-4.1
🔋 Status: ❌ Not working
📊 Samples: 4
🏷️ Sample IDs: 'site1', 'site2', 'site3', 'site4'
Query expected cell types and actual markers#
Let’s start by querying expected cell types and their markers in this tissue. We can use this step as a validation, to see how much the model “knows” about our data.
cell_ann.get_expected_cell_type_markers()
INFO Querying cell types.
INFO Writing expected cell types to `self.expected_cell_types`
INFO Querying cell type markers.
INFO Writing expected marker genes to `self.expected_marker_genes`.
INFO Filtering marker genes to only include those present in adata.var_names.
INFO Filtered 4 marker genes and removed 0 cell types with no marker genes left after filtering.
Let’s take a look at these.
cell_ann.expected_marker_genes
{'Hematopoietic stem cells (HSCs)': ['CD34',
'BAALC',
'GATA2',
'SPI1',
'MEIS1'],
'Multipotent progenitor cells (MPPs)': ['CD34',
'BAALC',
'MEIS1',
'GATA2',
'SPI1'],
'Common myeloid progenitors (CMPs)': ['CD34',
'GATA2',
'SPI1',
'MEIS1',
'RUNX1'],
'Common lymphoid progenitors (CLPs)': ['CD34',
'BAALC',
'MEIS1',
'GATA3',
'IL7R'],
'Megakaryocyte-erythroid progenitors (MEPs)': ['GATA1',
'GATA2',
'SPI1',
'CD34',
'MEIS1'],
'Granulocyte-monocyte progenitors (GMPs)': ['CD34',
'SPI1',
'GATA2',
'CEBPA',
'CSF1R'],
'Monocytes': ['CD14', 'LYZ', 'FCGR2A', 'CSF1R', 'SPI1'],
'Macrophages': ['CD163', 'CSF1R', 'SPI1', 'CD14', 'CD68'],
'Neutrophils (precursors and mature)': ['S100A8',
'S100A9',
'CSF3R',
'MPO',
'PRTN3'],
'Eosinophils (precursors and mature)': ['PRG2', 'GATA2', 'CEBPA', 'IL5RA'],
'Basophils (precursors and mature)': ['GATA2', 'IL3RA', 'CEBPA', 'FCER1G'],
'Erythroblasts': ['GYPA', 'HBB', 'HBA2', 'ALAS2', 'KLF1'],
'Pro-erythroblasts': ['GYPA', 'HBB', 'ALAS2', 'KLF1', 'CD34'],
'Normoblasts (late erythroblasts)': ['HBB', 'HBA2', 'GYPA', 'ALAS2', 'KLF1'],
'B cells (early and mature)': ['CD19', 'CD79B', 'MS4A1', 'POU2F2', 'SPI1'],
'T cells (mature, some precursors)': ['CD3E', 'CD3D', 'CD3G', 'CD2', 'CD7'],
'Natural killer (NK) cells': ['KLRK1', 'KLRD1', 'GNLY', 'GZMB', 'NKG7'],
'Plasma cells': ['XBP1', 'MZB1', 'IGHG1', 'IGKC', 'JCHAIN'],
'Dendritic cells (conventional and plasmacytoid)': ['CD86',
'ITGAX',
'FCER1G',
'CLEC4C',
'SPI1'],
'Mesenchymal stromal cells': ['VIM', 'CD44', 'ENG', 'VCAN'],
'Endothelial precursor cells': ['PECAM1', 'CD34', 'ENG', 'FLI1'],
'Mast cells (precursors and some mature forms)': ['KIT', 'FCER1G', 'GATA2']}
Ok, that looks reasonable. We could modify these expected cell types and marker genes by providing a dictionary and overwriting CellAnnotator.expected_marker_genes, thereby infusing prior knowledge. This information will be used later on to guide cell type label querying.
In the next step, we’ll compute marker genes for the clusters in our dataset.
cell_ann.get_cluster_markers()
INFO Iterating over samples to compute cluster marker genes.
We can also take a look at these marker genes. Under the hood, we created once cell_annotator.SampleAnnotator class per sample, and marker genes are written into these. Let’s take a look at an example:
cell_ann.sample_annotators["site3"].marker_gene_dfs["0"].head()
| gene | specificity | auc | |
|---|---|---|---|
| 0 | BANK1 | 0.803347 | 0.969336 |
| 1 | RALGPS2 | 0.754415 | 0.943715 |
| 2 | OSBPL10 | 0.932700 | 0.881645 |
| 3 | MS4A1 | 0.917240 | 0.880816 |
| 4 | EBF1 | 0.842919 | 0.845968 |
These are the top marker genes for one cluster in one sample.
Annotate clusters#
Using the marker genes computed above, we can now query the model for cell type labels. The expected cell types and marker genes queried above help to make predictions more robust and consistent across samples.
cell_ann.annotate_clusters()
INFO Iterating over samples to annotate clusters.
WARNING Not enough markers provided for cluster 0 in sample `site1` (0<2).
WARNING Not enough markers provided for cluster 10 in sample `site1` (1<2).
WARNING Not enough markers provided for cluster 18 in sample `site1` (0<2).
WARNING Not enough markers provided for cluster 8 in sample `site3` (0<2).
WARNING Not enough markers provided for cluster 14 in sample `site4` (1<2).
INFO Querying cell-type label de-duplication.
INFO Removed 15/50 cell types.
INFO Iterating over samples to harmonize cell type annotations.
WARNING Invalid global cell type name: Hematopoietic stem/progenitor cell (HSC/MPP) in sample `site2`. Re-using
the original name: Hematopoietic stem/progenitor cell (HSC/MPP).
INFO Writing updated cluster labels to `adata.obs[`cell_type_predicted'].
🧬 CellAnnotator
================
📋 Species: human
🔬 Tissue: bone marrow mononuclear cells
⏳ Stage: adult
🔗 Cluster key: leiden_shuffled
🔬 Sample key: Site
🤖 Provider: openai
🧠 Model: gpt-4.1
🔋 Status: ❌ Not working
📊 Samples: 4
🏷️ Sample IDs: 'site1', 'site2', 'site3', 'site4'
Within each cell_annotator.SampleAnnotator, we can inspect annotation results:
cell_ann.sample_annotators["site3"].annotation_df.head(10)
| n_cells | marker_genes | reason_for_failure | marker_gene_description | cell_type | cell_state | annotation_confidence | reason_for_confidence_estimate | cell_type_harmonized | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | 455 | BANK1, RALGPS2, OSBPL10, MS4A1, EBF1, BLK, PAX5 | None | The markers MS4A1 (CD20), PAX5, EBF1, and BLK ... | B cell | Normal | High | Multiple highly specific and canonical B cell ... | B cell |
| 1 | 356 | NFIA, SLC25A21, KIAA1211, ANK1, MBOAT2, CDC42B... | None | Several markers in cluster 1, including ANK1 a... | Early erythroid progenitor | Normal | Medium | While several markers overlap with erythroid l... | Early erythroid progenitor |
| 2 | 213 | SLC4A1, TSPAN5, ANK1, SLC25A37, SOX6, SPTA1, M... | None | The cluster markers SLC4A1, ANK1, SLC25A37, SO... | Normoblast (late erythroblast) | Normal | High | Markers are classic and highly specific for er... | Normoblast (late erythroblast) |
| 3 | 258 | ZNF385D, XACT, KIAA1211, MED12L, TAFA2, RYR3, ... | None | The markers for this cluster (ZNF385D, XACT, K... | Unknown | Normal | Low | None of the provided markers are standard for ... | Unknown |
| 4 | 353 | TCF7L2, MTSS1, WARS, PECAM1, MS4A7, RNF144B, C... | None | PECAM1 (CD31) is a classic marker for endothel... | Endothelial precursor cells | Normal | High | PECAM1 is highly specific for endothelial cell... | Endothelial precursor cells |
| 5 | 38 | IFNG-AS1, MZB1, TXNDC5, DENND5B, SEL1L3, AL589... | None | Cluster 5 expresses MZB1 and IFNG-AS1, both of... | Plasma cells | Normal | High | Key plasma cell marker MZB1 is present, suppor... | Plasma cells |
| 6 | 248 | NEGR1, CST3, CCSER1, RUNX2, HLA-DPB1, FLT3, CIITA | None | RUNX2 is a transcription factor that can be se... | Multipotent progenitor (MPP)/early dendritic-l... | Normal | Medium | Marker overlap with both lymphoid/dendritic pr... | Multipotent progenitor (MPP)/early dendritic-l... |
| 7 | 623 | APP, AC023590.1, RUNX2, FAM160A1, PLXNA4, CUX2... | None | The marker genes include APP (amyloid beta pre... | Unknown | Normal | Low | None of the marker genes are classical markers... | Unknown |
| 8 | 129 | Not enough markers provided for cluster 8 in s... | Unknown | Unknown | Unknown | Unknown | Unknown | Unknown | |
| 9 | 581 | CAMK4, BCL11B, INPP4B, LEF1, ANK3, IL7R, MLLT3 | None | CAMK4, BCL11B, LEF1, and IL7R are canonical ma... | T cells (early/precursor) | Normal | High | The presence of multiple key T-lineage specify... | T cell (early precursor or thymocyte) |
You can see that the model returns Unknown when the provided information isn’t enough to output a reliable prediction.
Annotations have been automatically harmonized across samples (cell_type_harmonized) and written to the underling anndata.AnnData object, by default to cell_type_predicted.
Evaluate#
First, let’s get clusters into a consistent ordering across annotations. This will make it easier to interpret the results in a confusion matrix.
obr = ObsBeautifier(adata, model="o4-mini") # reasoning models work best here
# bring categories into a more meaningful order
obr.reorder_categories(keys=["cell_type", "cell_type_predicted"])
# assign colors to the categories
obr.assign_colors(keys=["cell_type", "cell_type_predicted"])
INFO ✅ OPENAI API key is available
INFO Querying label ordering.
INFO Reordering categories for key 'cell_type'
INFO Reordering categories for key 'cell_type_predicted'
INFO Querying cluster colors.
INFO Assigning colors for key 'cell_type'
INFO Assigning colors for key 'cell_type_predicted'
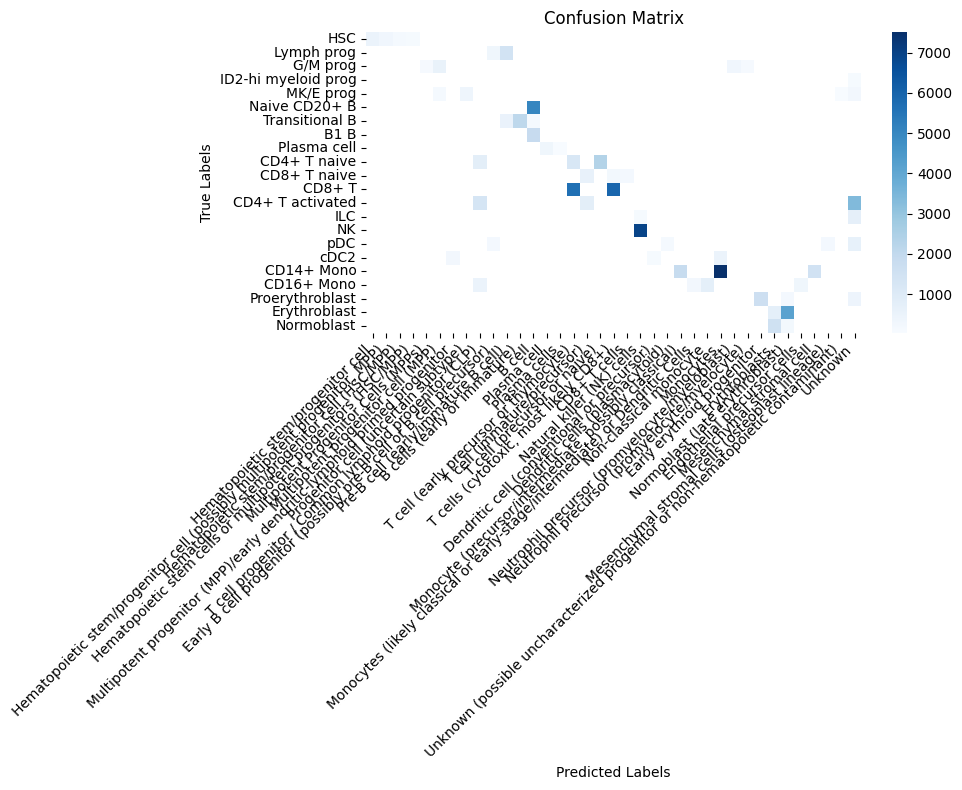
Compute ground-truth with predicted cell type labels in a confusion matrix (across all samples).
df = adata.obs.groupby(["cell_type", "cell_type_predicted"], observed=True).size().unstack()
# Plot the heatmap
plt.figure(figsize=(10, 8))
sns.heatmap(df, annot=False, cmap="Blues", xticklabels=True, yticklabels=True)
plt.title("Confusion Matrix")
plt.xlabel("Predicted Labels")
plt.ylabel("True Labels")
plt.xticks(rotation=45, ha="right")
plt.yticks(rotation=0)
plt.tight_layout()
plt.show()
We can also compare ground-truth and predicted cell types in the UMAP embedding.
sc.pl.embedding(adata, basis="umap", color=["cell_type", "cell_type_predicted"], wspace=0.6)